通过biomod2整合多个模型对物种进行预测——白芷为例
前准备
项目文件结构
1 | Angelica_BIOMOD2/ |
renv是 R 的项目环境管理工具,用于固定项目依赖包的版本,确保代码可复现。renv::init()用于初始化项目环境
1 | install.packages("renv") |
1. 环境准备
1 | renv::install(c("biomod2", "terra", "sp", "dismo", "ggplot2", "viridis",'tidyterra')) |
2. 数据导入
白芷分布点数据(presence only) CSV格式示例:
1 | species,lon,lat |
- 读取 CSV 格式的分布点数据
- 将数据转换为空间点对象(sp包格式),指定经度和纬度为坐标
- 设置坐标参考系(CRS)为 WGS84(经纬度坐标系,全球通用)
1 | occ <- read.csv("data/Angelica_points.csv") |
- 读取
env文件夹下所有.tif格式的环境变量文件(如 WorldClim 的 19 个生物气候变量、HWSD2 土壤变量等) - 将多个环境变量合并为一个raster栈(
env_stack),方便后续统一处理 - 打印环境变量名称,确认数据正确导入
- 绘制第一个环境变量(如
Bio1,年平均温度)的空间分布,并叠加白芷分布点(红色小点),直观展示分布点与环境的关系
1 | env_files <- list.files("env/", pattern = ".tif$", full.names = TRUE) |
3. 格式化输入数据
BIOMOD_FormatingData是BIOMOD2的核心函数,用于将分布数据和环境数据格式化为模型输入格式- 因原始数据只有存在点(presence-only),需生成伪缺失点(Pseudo-Absences, PA)作为模型的 “缺失” 样本(模型训练需要存在 / 缺失对比)
- 这里设置生成
3组伪缺失点,每组10000个,随机分布在研究区域
1 | biomod_data <- BIOMOD_FormatingData( |
4. 模型配置
- 定义 5 种常用物种分布模型的参数:
GLM(广义线性模型):使用二次项,无交互项,以AIC准则选择变量GAM(广义可加模型):平滑参数k=4,二项分布链接函数RF(随机森林):500棵决策树GBM(梯度提升机):3000棵树,学习率0.005,控制过拟合MAXENT(最大熵模型):包含线性项、二次项、hinge项等特征
user.val整合所有模型的参数,allModels指定最终运行的模型列表
1 | # 定义模型参数 |
- 用
bm_ModelingOptions统一配置建模参数,指定数据类型、模型列表、参数来源等,为后续建模做准备。
1 | myOptions <- bm_ModelingOptions( |
5. 运行建模
BIOMOD_Modeling是模型训练的核心函数,基于输入数据和配置运行所有模型- 交叉验证(
10次重复,75%训练 /25%验证)用于评估模型稳定性 - 记录
TSS和ROC作为模型性能指标,并计算变量重要性(评估环境变量对分布的影响程度)
1 | biomod_model_out <- BIOMOD_Modeling( |
6. 模型评估
- 提取模型评估结果(如各模型在训练集和验证集上的
TSS、ROC值),用于判断模型表现(TSS>0.7、ROC>0.8通常认为模型较好)
1 | evals <- get_evaluations(biomod_model_out) |
- 可视化模型评估结果:
- 平均评估图:展示各模型在训练 / 验证集上的平均性能;
- 箱线图:展示不同模型在多次重复中的性能分布,反映稳定性。
1 | bm_PlotEvalMean(bm.out = biomod_model_out, dataset = 'calibration') |
7. 变量重要性
- 绘制变量重要性箱线图,展示不同环境变量在各模型(
algo)和多次重复(run)中的重要性分布 - 帮助识别对白芷分布影响最大的环境变量(如某个气候因子或土壤因子)
1 | bm_PlotVarImpBoxplot(bm.out = biomod_model_out, group.by = c('expl.var', 'algo', 'algo')) |
8. 集成建模
- 集成多个单个模型的结果,减少单一模型的不确定性,提高预测可靠性
- 先筛选出性能较好的模型(
TSS≥0.7),再通过 3 种算法(平均、交叉验证平均、加权平均)生成集成模型
1 | biomod_em <- BIOMOD_EnsembleModeling( |
9. 集成预测
BIOMOD_Projection:用单个模型基于当前环境变量(env_stack)预测白芷的适宜性分布;BIOMOD_EnsembleForecasting:基于集成模型生成最终的适宜性预测结果(综合多个模型的优势)。
1 | biomod_proj <- BIOMOD_Projection( |
1 | plot(biomod_em_proj) # 快速可视化集成预测结果 |
10. 结果导出
1 | output_path <- paste0(getwd(), "/outputs/Angelica_dahurica/proj_current/") |
11. 可视化预测图
1 | pred_raster <- raster(paste0(output_path, "Angelica_EMmeanByTSS.tif")) |
最终效果: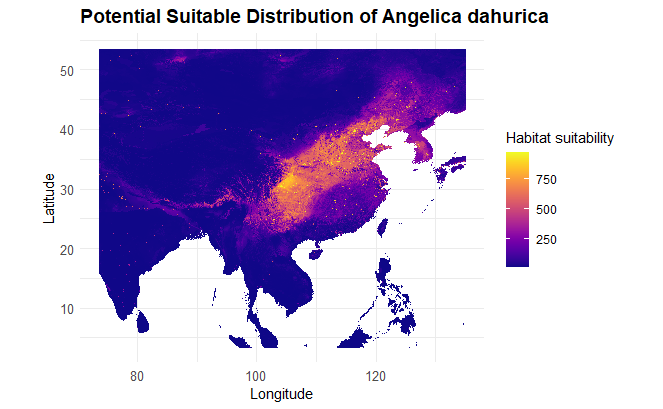
- 标题: 通过biomod2整合多个模型对物种进行预测——白芷为例
- 作者: Riceneeder
- 创建于 : 2025-10-30 17:54:35
- 更新于 : 2025-11-28 16:40:49
- 链接: https://gankun.cn.lu/posts/2025-10-30/
- 版权声明: 版权所有 © Riceneeder,禁止转载。
评论